
Welcome to the NFINtb Nothobranchius furzeri transcriptome browser
The NFINtb Nothobranchius furzeri Information Network transcriptome browser is a database of cDNA contig assemblies of the short-lived teleost fish Nothobranchius furzeri supplemented with the best currently available annotation.
Content:
- Introduction
- Start page
- Statistics page
- Query page
- Transcript table page
- Sequence page
- Blast page
- GO page
- Help page
- Contact page
- Methods table
- Data resources
- How to cite
1. Introduction
The NFINtb Nothobranchius furzeri transcriptome browser provides access to the annotated transcript catalogue, including detailed BLAST results, predicted protein domains, associated gene ontology terms, microsatellites and gene expression data. Further information includes sequenced libraries, distribution of contig lengths and annotation numbers, respective GO terms and corresponding transcript contigs is given. A query mask allows keyword (e.g. annotation, gene symbol, GO category) and BLAST searches by sequence similarity. N. furzeri sequences can be retrieved by batch download including annotation details.
2. Start page
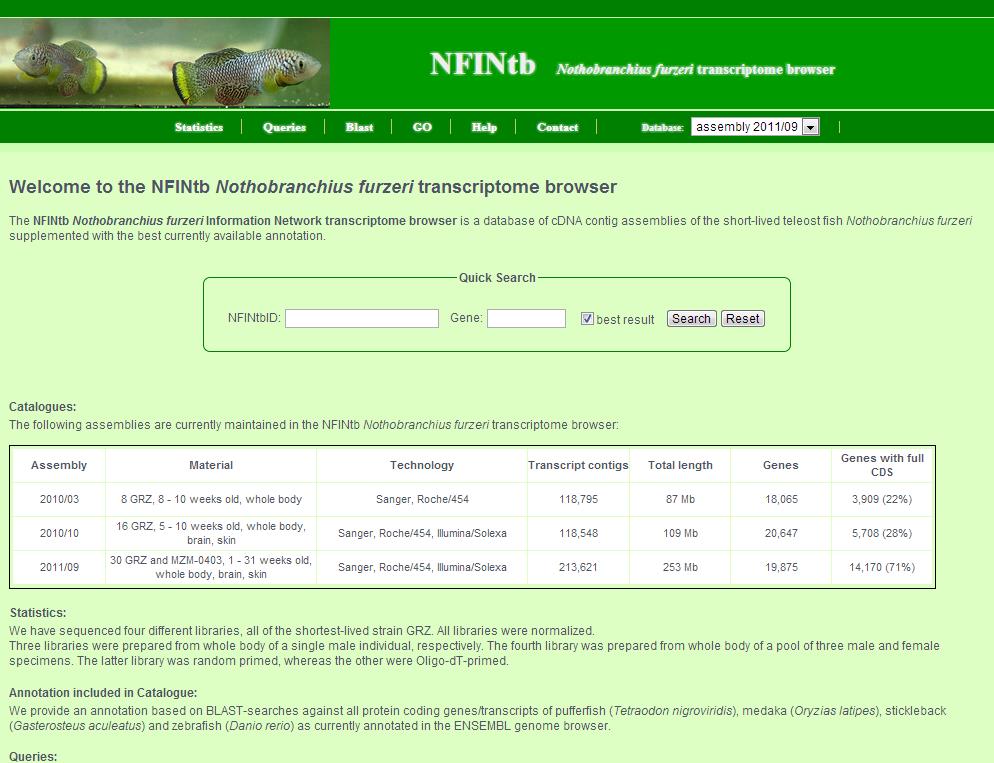
1. First, choose the catalogue of interest. A table of all assembled catalogues is given further below. 2. Then, you can click on several links of the link bar to get the information you are interested in. A more comprehensive explanation of each pages follows. 3. Last, there is also a quick search field where you can search either by NFINtbID or by gene symbol. If you search by gene symbol, mark the check box to get only the best (longest) transcript contig per gene.
3. Statistics page
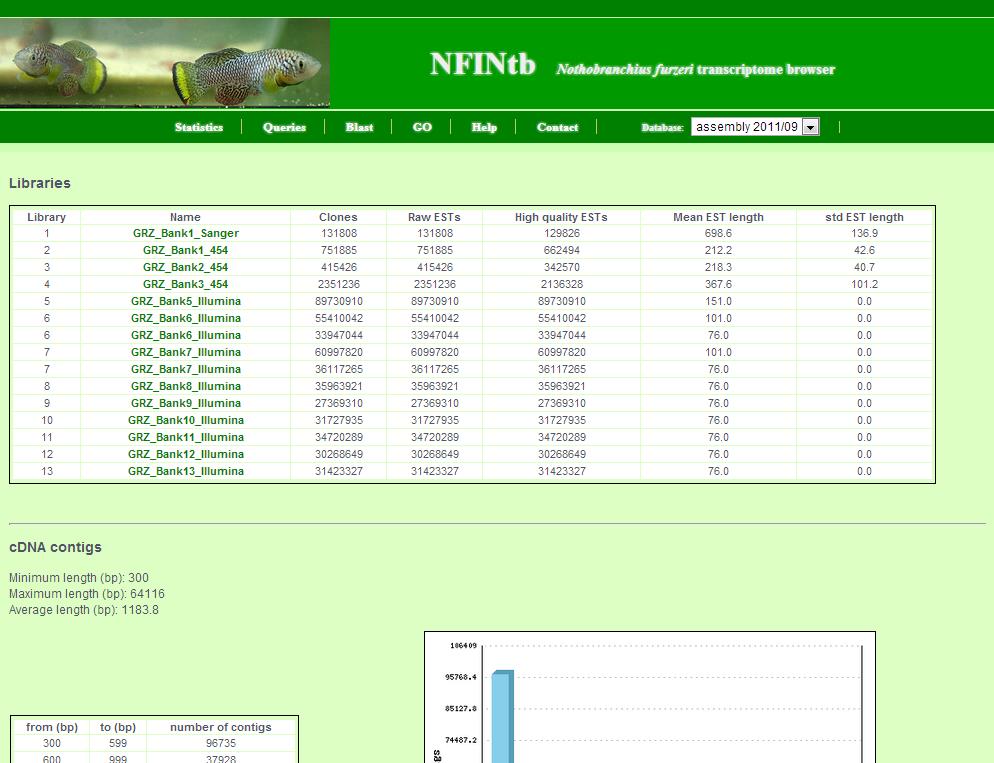
The statistics page shows general information about the transcript catalogue.
This includes:
- Information about assembled sequencing data and libraries
- Transcript contig length distribution
- BLAST, protein domain prediction and annotation numbers
- Detected SNP (single nucleotid polymorphism) and SSR (simple sequence repeats)
- Information about possible contamination
4. Query page
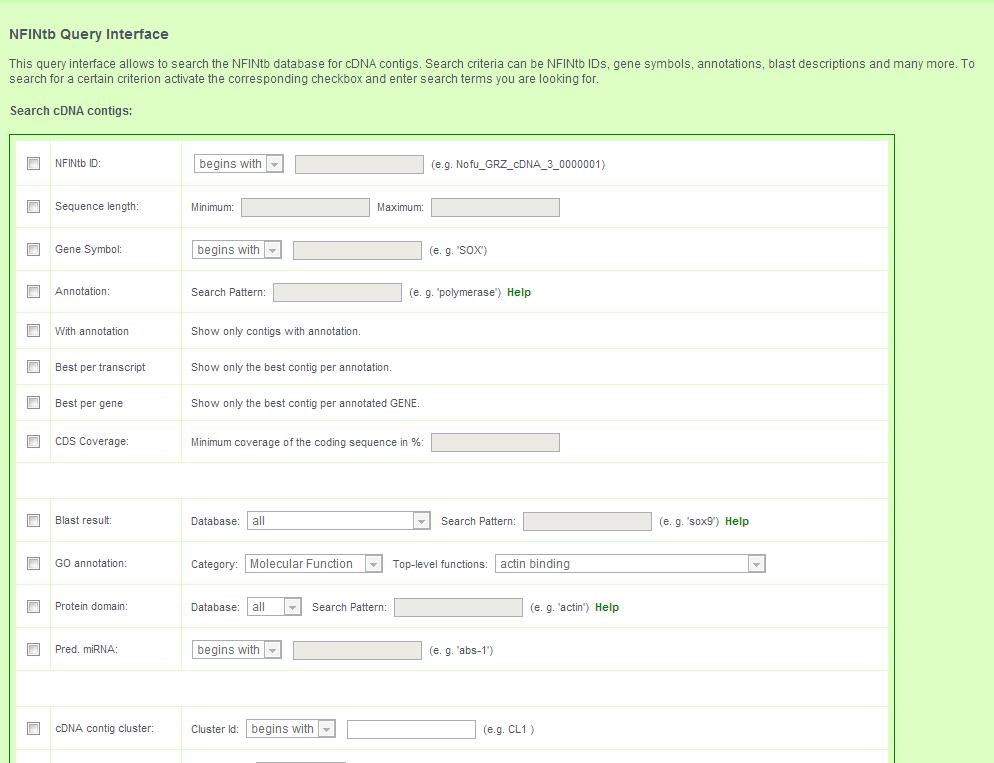
The query page provides an interface for complex queries to search the transcript catalogue.
4.1 Transcript contigs can be searched by:
- NFINtbID
- Minimum and maximum length
- Gene symbol
- Annotation (Text search)
- Annotation status - yes/no
- Show only the best transcript contig per annotated protein
- Show only the best transcript contig per annotated gene
- Minimum coding sequence coverage in percent
- Search BLAST results (Text search)
- GOslim annotation in one of the three ontologies/descriptions
- Search protein domain predictions (Text search)
- Predicted miRNA homology
- SNP ID
- SNP type
- SSR ID or type
- Contamination status
Fields marked with 'Text search' allow some advanced search strategies like wild cards and pattern grouping as well as some logical operators. Rules are explained below:
4.2 Text Search means:
- the search is always case insensitive
- all words must be larger than 3 characters
- words that are too common like 'the' or 'some' are ignored
- all words are matched completely
- use '*' for incomplete matches (Example: Pol*)
- use search operators for inexact searches
4.3 Search Operators include:
- +
- A leading plus sign indicates that this word MUST be present in a result. Example: +protein +kinase
- -
- A leading minus sign indicates that the word MUST NOT be present in a result. Example: +protease -kinase
- ( )
- Parentheses group words to look for several possibilities. Example: (RNA|DNA) polymerase
- *
- Use the '*' operator to look for truncated words. Example: Protea*
- " "
- A phrase that is enclosed within double quote (") characters matches
only results that contain the phrase literally. Example: "this is a example"
5. Transcript table page
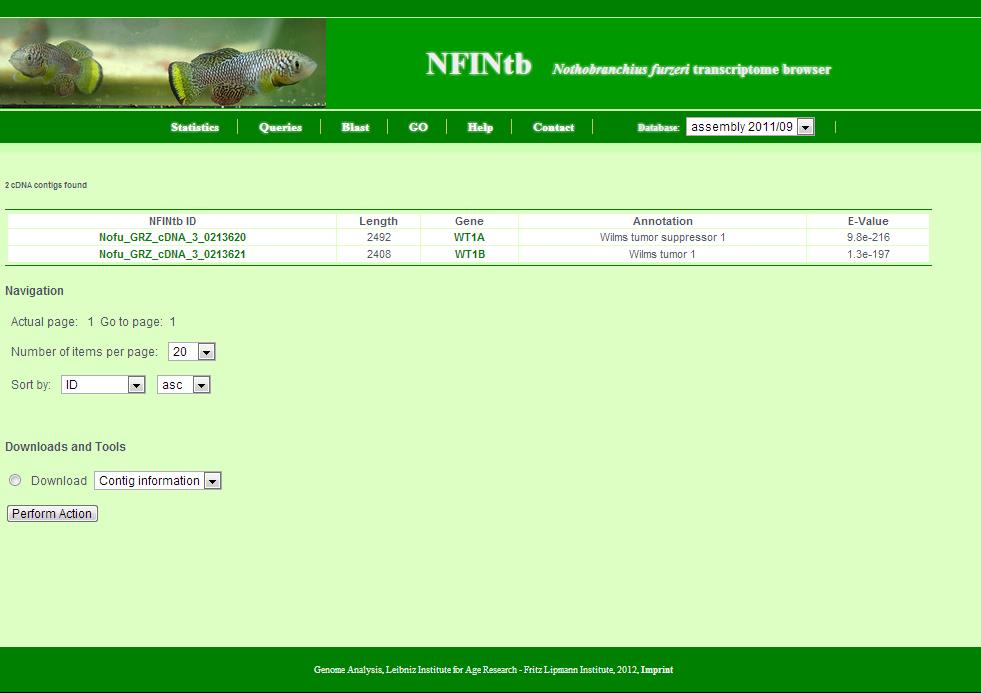
This page appears summarises the results of a search. Generally, it shows a number of transcript contigs as a table and provides tools to batch retrieve these.
The page is composed as follows:
- The number of retrieved transcript contigs.
- The table with transcript contigs: Each row contains NFINtbID, length, gene symbol, annotation and e-value of annotation. NFINtbIDs are linked to individual transcript contig pages. Gene symbols are linked to the NCBI Gene database. Also, as a side remark, e-values of unannoated transcript contigs are set to 1 for technical reasons.
- The navigation allows you to go the next page, set the number of transcript contigs per page and change the order of transcript contigs listed in the table. Sort criteria are NFINtbID, length, gene, annotation and e-value. 'Asc' means ascending and 'desc' means descending.
- Tools allow you to retrieve the set of transcript contigs as sequence file in fasta format or list file in csv format. Further (yet inactivated) tools include the download of gene expression data, the download of corresponding zebrafish, medaka, stickleback and tetraodon orthologs and some basic functional gene enrichment analysis via babelomics.
6. Sequence page
7. Blast page
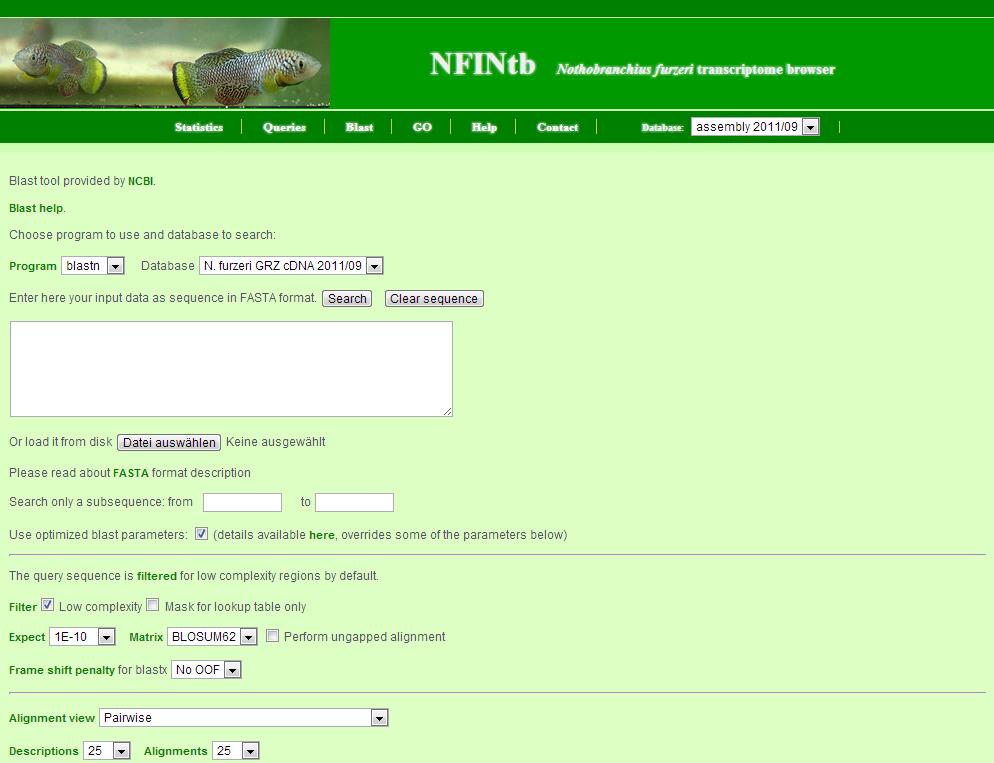
The BLAST server allows you to do similarity searches to find transcript contigs of interest.
Here are some general remarks for its use:
- the query sequence can either be copy-and-pasted or uploaded as a file
- the search can be restricted to a subsequence of the query
- the BLAST server also contains parameter sets to optimize different searches - it will choose the approbiate set depending on your input
- additional parameters can be set below - but will be overridden by the optimised parameter sets above
- the search can be made more stringent by choosing a smaller e-value (Expect)
- the output is in pairwise alignment but other formats are also available
- to import the results in excel, choose the format "Hit Table" and load the table as a CSV file
- limit the number of hits and alignments to avoid large blast reports
- IMPORTANT: this is only a small-scale BLAST server - please blast only one sequence at a time - longer searches will be terminated by timeout
8. GO page
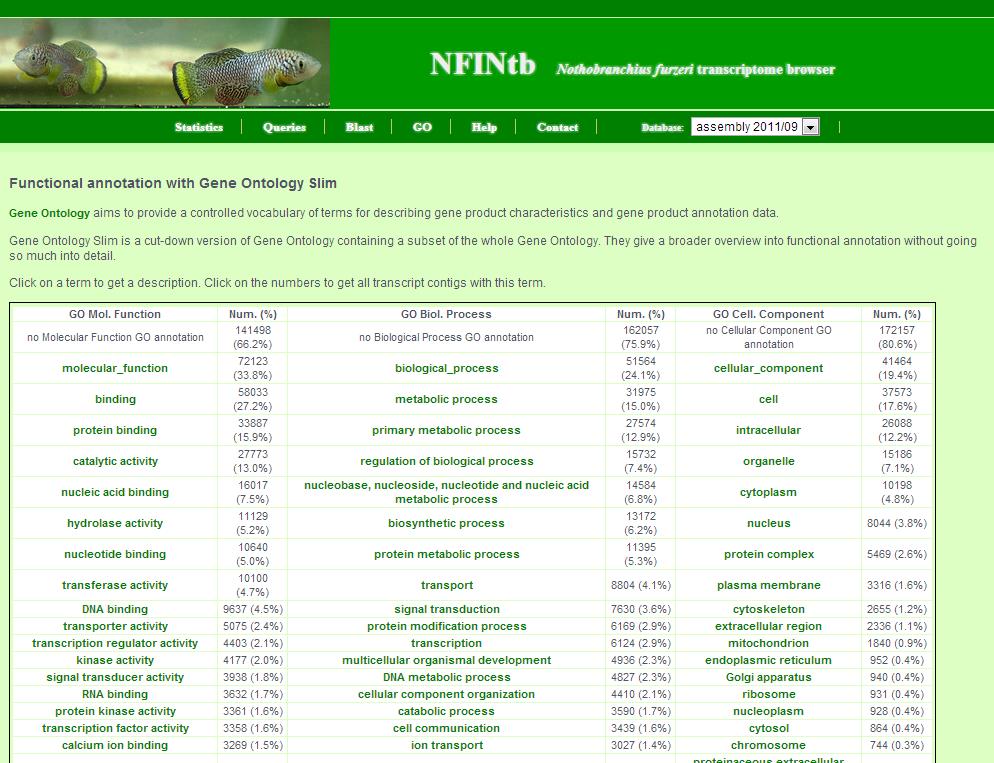
The GO page
9. Help page
10. Contact page
8. Methods table
| Task | Program | Version | Arguments | Description |
| 1. Read processing | ||||
| Sanger quality clipping | Lucy | 1.19p | -error 0.02 0.015 -bracket 10 0.015 -window 50 0.08 10 0.3 -vector vector_db insert_sites | Trims low-quality regions and remove remaining vector sequences in Sanger reads. |
| Sanger and 454/Roche vector/contaminant/poly(A) removal | SeqClean | 1.0 | -n 5000 -l 80 -v vector_db -s contaminant_db -N -L | Trims additional vector and poly (A) tails in Sanger and 454/Roche reads, and removes contaminant reads. |
| Sanger and 454/Roche low-complexity masking | SeqClean | 1.0 | -n 5000 -l 80 -A -M | Masks low-complexity repeats in Sanger and 454/Roche reads. |
| Sanger and 454/Roche repeat masking | RepeatMasker | 3.2.9 | -norna -xsmall -noint -qq -gccalc -lib nfurzeri_repeats | Masks complex repeats in Sanger and 454/Roche reads. |
| Solexa/Illumina read filtering | SGA | 0.9.12 | sga preprocess --phred64 -p 1 -f 20 --dust | Filters Solexa/Illumina reads for quality and low complexity. |
| Solexa/Illumina error correction | SGA | 0.9.12 | sga index reads;sga correct reads; | Corrects obvious sequencing errors in Solexa/Illumina reads . |
| Solexa/Illumina duplicate read removal | SGA | 0.9.12 | sga index reads;sga filter --no-kmer-check reads; | Removes exact duplicates in Solexa/Illumina reads. |
| 2. Assembly | ||||
| Sanger and 454/Roche assembly | PAVE | 2.0 | default cfg file | Assembly of Sanger and 454/Roche reads |
| Solexa/Illumina reads, PAVE contigs assembly | CLC Assembly Cell | 3.2.2 | -m 300 -p fb se 0 10000 | Assembly of Solexa/Illumina reads onto PAVE contigs |
| Solexa/Illumina/PAVE contigs reassembly | TGICL | 2.0 | -l 100 -O '-d 30000 -f 5000 -p 90' | Extends and joins contigs with relaxed parameters. |
| Solexa/Illumina/PAVE - removal of redundant contigs | CD-HIT-EST | 4.5.5 | -T 8 -c 0.99 -aS 0.9 -n 10 -B 1 -M 0 | Removes highly redundant/overlapping contigs. |
| 3. Annotation | ||||
| BLAST Contamination screen | WU-tBLASTx | 2.0MP-WashU | wordmask=seg W=4 T=999 hitdist=40 nogap V=25 B=25 | Transcript contigs are compared against a contamination database. |
| BLAST annotation | WU-BLASTx,WU-BLASTn,WU-tBLASTx | 2.0MP-WashU | wordmask=seg lcmask W=4 T=20 (protein);M=1 N=-3 Q=3 R=3 W=15 wordmask=seg lcmask (nucleotide) | Transcript contigs are compared against several protein and nucleotide database for annotation. |
| Open reading frame (ORF) prediction | prot4EST | 2.0 | BLASTx against NCBI nr, ESTScan with N.furzeri matrix, longest ORF | Determines ORF in the transcript contigs that are translated to proteins. |
| Protein domain prediction | HMMER | 3.0 | default parameters | Searches translated ORF for conserved protein domains. |
| Protein domain prediction | HMMER | 3.0 | default parameters | Searches translated ORF for conserved protein domains. |
| Orthologs in other fish species | WU-BLASTx,WU-tBLASTn | 2.0MP-WashU | wordmask=seg lcmask W=4 T=20;wordmask=seg W=4 T=20 | Transcript contigs are compared against proteins of other fish. Fish proteins are compared against transcript contigs. Best bidirectional hits are calculated. |
| Detection of paralogs | WU-BLASTx | 2.0MP-WashU | wordmask=seg lcmask W=4 T=20 | Transcript contigs are compared against other fish's proteins. The associated gene symbols of the proteins are used to fetch |
9. Data resources
Sequencing data and assembly are online available in different databases. The NCBI BioProject page "Sequencing and assembly of the Nothobranchius furzeri transcriptome" (PRJNA85613) provides a good overview and starting point. 454/Roche and Solexa/Illumina data is also summarised in the NCBI SRA, study acession SRP010930.
Individual libraries in public databases:
- Sanger libraries, NCBI dbEST:
- JZ200028 - JZ330399, please note that dbEST only accepts processed high-quality sequences (no raw data), therefore the number of submitted sequences is lower than given in the manuscript's Table
- 454/Roche libraries, NCBI SRA:
- Solexa/Illumina libraries, NCBI SRA:
Transcriptome Shotgun Assembly (TSA)/GenBank:
- assembly 2011/09: GAIB01000000 (GAIB01000001 - GAIB01210031)
11. How to cite
Please cite the following paper if you use data from this browser:
Petzold A, Reichwald K, Hartmann N, Groth M, Taudien S, Shagin D, Priebe S, Englert C, Platzer M:
The transcript catalogue of the short-lived fish Nothobranchius furzeri provides insights into age-dependent changes of mRNA levels.
BMC Genomics 2013, 14:185. doi: 10.1186/1471-2164-14-185.
(http://www.ncbi.nlm.nih.gov/pubmed/23496936)
|
|
Beutenbergstraße 11 D-07745 Jena • Germany |
Phone: +49 3641 65-6000 Fax: +49 3641 65-6351 |
E-mail:
info@leibniz-fli.de
www.leibniz-fli.de |
Imprint
Data Privacy |

|